
SaMD – USA and EU Approach Differences
As per the FDA, the term Software as Medical Device (SaMD) is defined as “software intended to be used for one or more medical purposes that perform these purposes without being part of a hardware medical device.
Any software that helps diagnose, track, monitor, or evaluate a medical condition or state is referred to as SaMD and is not a part of the hardware device.
Examples of SaMD
- A mobile application for imaging MRI on your Mobile Phone
- Sleep detecting application that displays data and results on the mobile phone
- Software clone of the digital mammogram machine used during the breast cancer detection
- Software for monitoring the pacemaker activity on your mobile phone
Key Characteristics
- Fast feedback loop can enable quicker product literation’s.
- Improved health outcomes powered by data.
- Collect large amounts of data quickly.
- Easily solicit user feedback through its availability.
- Shorten time to market, and drive faster innovation.
- Faster production and feedback to drive faster innovation
SaMD Classification
SaMD Risk Class and “Levels of Concern” in the US:
FDA classifies SaMD using the same risk classes as it does for traditional medical devices: Class I, Class II, and Class III.
Selecting a “level of concern” for your pre-market application to the FDA does not establish your risk class. The level of concern simply indicates the type of documentation required for your pre-market submission. While it may be strongly connected to risk class, your level of concern is not used to establish your device’s risk class.
Medical Device Software Risk Class and “Rule 11” in the EU:
As with the US, there is no MDSW specific risk classification in the EU. Medical device software uses the same risk classification as traditional medical devices: class I, class IIa, class IIb, and class III.
But the EU MDR has an outline for how you should go about determining your medical device software risk class, identified as Rule 11.
Rule 11, which can be found in Annex VIII of EU MDR, states:
Software intended to provide information that is used to take decisions with diagnosis or therapeutic purposes is classified as class IIa, except if such decisions have an impact that may cause:
- Death or an irreversible deterioration of a person’s state of health, in which case it is in class III; or
- A serious deterioration of a person’s state of health or a surgical intervention, in which case it is classified as class IIb.
Software intended to monitor physiological processes is classified as class IIa, except if it is intended for monitoring of vital physiological parameters, where the nature of variations of those parameters is such that it could result in immediate danger to the patient, in which case it is classified as class IIb. And all other software is classified as class I.
When does a SaMD require a new submission?
Making a change to a medical device that is on the market will always require scrutiny. At the very least, it needs to document the change that was made. But if a change is significant enough, it may require resubmitting the documentation needed to get the device on the market in the first place.
Making changes to SaMD in the US
Software as a medical device is similar to traditional medical devices in that if you want to make changes to your device, you have two options:
- Notify FDA of the change via a new 510(k), a special 510(k), or PMA supplement
- Document the change internally via a letter-to-file
The decision you make is based on the significance of the change and whether it affects the device’s safety, efficacy, or overall performance. For a hardware medical device, it is usually easy to determine whether your change is large enough to warrant informing the FDA. However, choosing software can be more difficult.
FDA has released guidance on Deciding When to Submit a 510(k) for a Software Change to an Existing Device to help SaMD manufacturers.
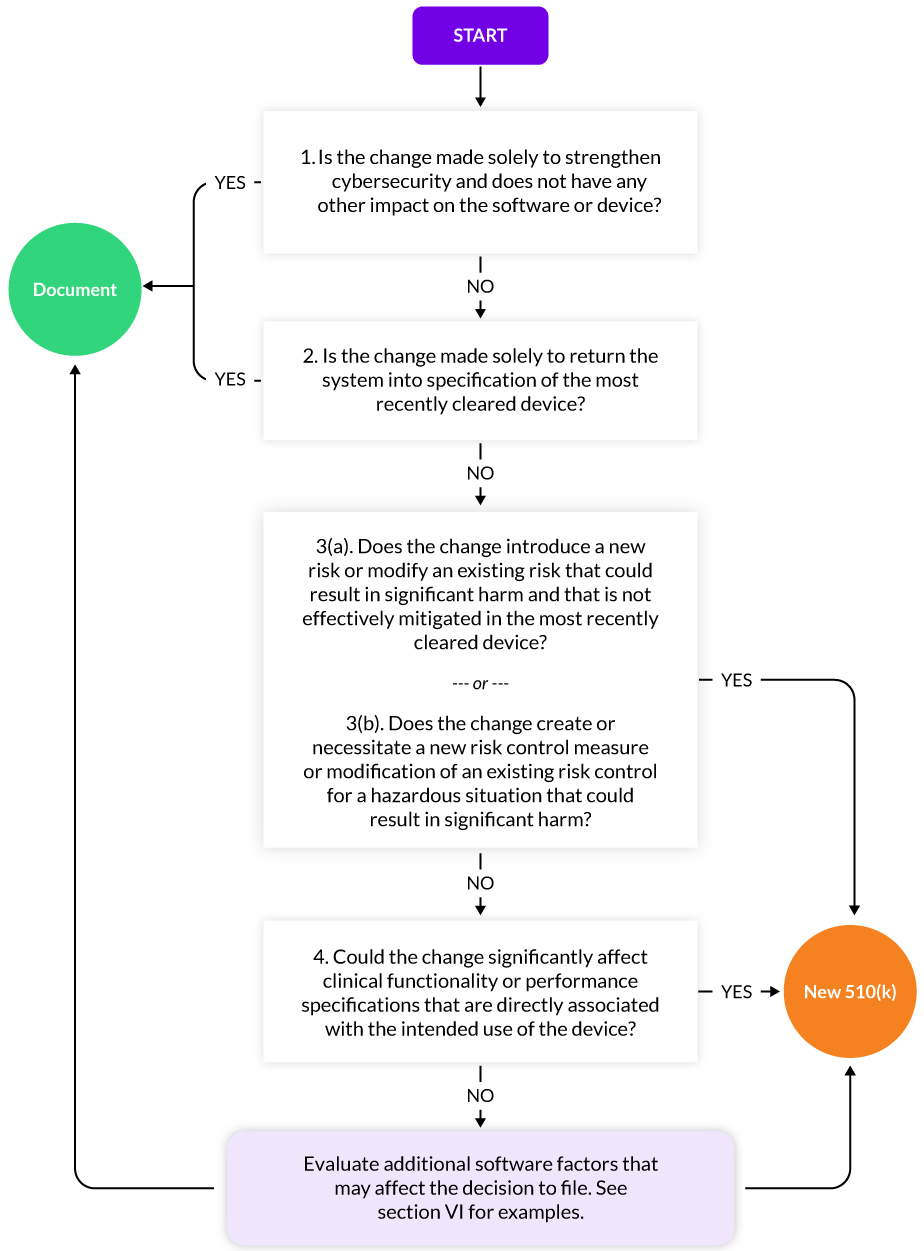
Making Changes to SaMD in the EU:
The requirements for a notification of a change in your device in the EU are similar. Annex X of EU MDR states:
Changes to the approved device, including limitations on its intended purpose and use conditions, must be approved by the notified body that issued the EU type-examination certificate if they affect conformity with the product’s general safety and performance requirements or the conditions prescribed for its use. The notified authority will review the proposed adjustments, notify the manufacturer of its decision, and give him a supplement to the EU type-examination report. Any modification to the approved type should be authorized as a supplement to the EU type examination certificate.
Although the MDR requirements are not exactly the same as the FDA guidelines for making changes to SaMD. Notice a similar theme running through both: modifications that change the intended use of the device require a new submission to the respective notified body.
Regulatory Requirements – FDA
The FDA has published many guidance documents on software regulation, specifically SaMD.
The FDA regulates some kinds of software as medical devices, while others are not. Additionally, certain software is subject to ‘enforcement discretion’, which means that the FDA may not aggressively pursue enforcement unless there is a compelling cause to do so.
Regulatory Requirements – EU
The EU uses the term ‘medical device software’ (MDSW) instead of SaMD. It defines MDSW as software that is intended to be used, alone or in combination, for a purpose as specified in the definition of ‘medical device’ in the MDR or IVDR.
The EU uses a different term because:
1. it does not regulate SaMD with functionality that is limited to storage, communication, lossless compression, or simple searching or that is intended for the benefit of populations rather than individuals and
2. Contrary to SaMD, software that fulfills a medical purpose but is also intended to drive or influence the use of a medical device is still considered to be MDSW, whereas, according to the IMDRF notes, SaMD cannot drive a medical device. Qualification as MDSW is regardless of:
- Its location – for example operating in the cloud, on a computer, on a mobile phone or as an additional functionality on a hardware medical device
- Whether the software, in addition, also drives or influences the use of a (hardware) medical device
Conclusion:
The regulatory landscapes for Software as a Medical Device (SaMD) in the European Union and the United States present notable differences rooted in their respective approaches to safety, innovation, and market access. The EU’s framework, with its emphasis on stringent conformity assessments and rigorous pre-market evaluations, prioritizes a high degree of patient safety and device efficacy, ensuring comprehensive oversight through the Medical Device Regulation (MDR) and In Vitro Diagnostic Regulation (IVDR). Conversely, the US approach, governed by the Food and Drug Administration (FDA), adopts a more flexible and expedited pathway for SaMD, fostering innovation and market entry through mechanisms such as the De Novo classification and software-specific guidance documents.
Get the latest updates from DDi
Explore Topics
Recent Blogs
 The Future of Regulatory Opera…Regulatory Automation
The Future of Regulatory Opera…Regulatory Automation How eIFU Enhances Compliance a…Regulations
How eIFU Enhances Compliance a…Regulations Global UDI updates in recent m…UDI
Global UDI updates in recent m…UDI



CONNECT WITH US

Let's talk about how DDi can help you